Students involved in our summer research program present their findings at both local and national American Chemical Society meetings. Some of the types of data collected and presented by students are depicted in Figure 3, and include optimized geometries, molecular orbitals, and potential energy surface scans.
Castro / Karney Research Group - Department of Chemistry
Computational organic chemistry with a focus on annulenes, tunneling, Möbius topology, and PAHs

The Castro/Karney research group uses computational tools to solve physical organic chemistry problems. Most of our research is carried out by undergraduate students, the majority of whom join our group after taking sophomore organic chemistry. Using state of the art software (e.g. Gaussian, Molcas, Molpro, Polyrate) we probe mechanisms of reactions that occur at both low and high temperatures, we study molecular properties, and we try to make predictions that will be useful to experimentalists. Much of our earlier work focused on annulenes and the role that Möbius-topology species play with regard to their reaction dynamics (Fig 1). We have also worked on elucidating the mechanisms for Stone-Wales rearrangements (SWRs) and other high-temperature transformations in carbon-rich materials (Fig 2). These latter reactions are relevant to soot formation during combustion, and to the annealing of fullerenes and carbon nanotubes. Current work involves probing the contribution of heavy-atom tunneling to pericyclic reactions in biosynthetic pathways and biomimetic syntheses. The National Science Foundation has funded our research since 2006.
Figure 1. Novel cis/trans isomerization reaction mechanism via Möbius π-bond shifting.
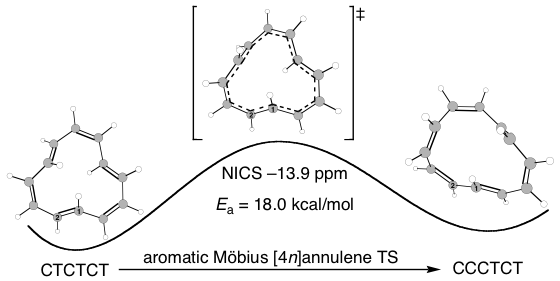
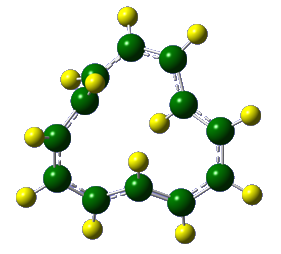
Figure 2. Representative Stone–Wales rearrangements in pentafulvalene and buckminsterfullerene.

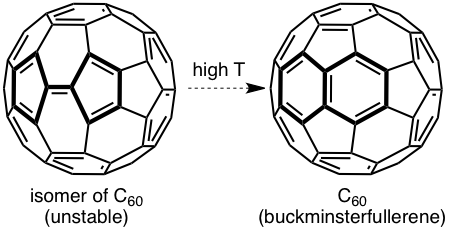

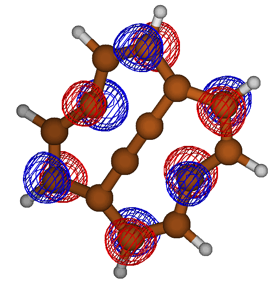
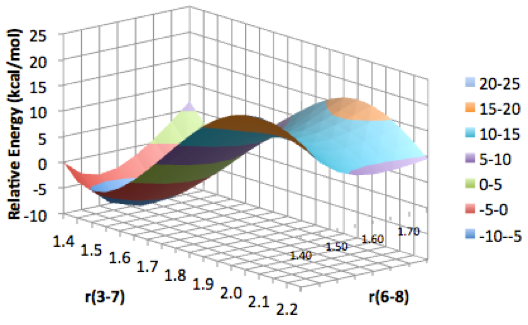
Figure 3. Examples of results obtained by students in the group. Clockwise from upper left: Optimized molecular structures, molecular orbitals, and two-dimensional potential energy surface scans.
Figure 3. Examples of results obtained by students in the group. Top to bottom: Optimized molecular structures, molecular orbitals, and two-dimensional potential energy surface scans.